Chapter 5: Pharmacokinetics
2nd edition as of August 2022
Chapter Overview
For the last three chapters, we have scrutinized the human nervous system from the overall structure down to the individual synapse. For the next two chapters, we will be bringing drugs to the forefront by exploring pharmacology, which can be divided into two main branches. The first branch—pharmacokinetics—is the focus of this chapter. The second branch, pharmacodynamics, will be covered in the next chapter.
Pharmacology (from pharmakon in Greek for “drug” and logia for “study of”) is often confused with pharmacy, and that assumption would be incorrect. Pharmacology is a biomedical science involving the discovery, research, and characterization of drugs and their actions and effects. Pharmacy is a direct-patient healthcare profession dealing with the restoration and maintenance of health in patients. Just like neuroscience, pharmacology is a vast and complex subject that is often studied over multiple semesters. This discussion will be simplified somewhat, but there will still be many new terms and basic concepts to learn. Nevertheless, doing so will be vital, as these ideas will show up again and again when we transition to looking at specific types of drugs for the rest of the semester. Stay focused, view the recommended videos, and be sure to reach out to your instructor or TA if you need help.
Chapter Outline
- 5.1.1. The Pharmaceutical Sciences
- 5.1.2. ADME
- 5.2.1. Determinants of Absorption
- 5.2.2. Enteral Routes
- 5.2.3. Parenteral Injection Routes
- 5.2.4. Parenteral Non-Injection Routes
- 5.3.1. Plasma Protein Binding
- 5.3.2. The Blood-Brain Barrier
- 5.4.1. Metabolism in the Liver
- 5.4.2. Enzyme Induction and Inhibition
- 5.4.3. Prodrugs
- 5.5.1. Routes of Excretion
- 5.5.2. Elimination Kinetics
Chapter Learning Outcomes
- Define pharmacology and describe the pharmaceutical sciences.
- Describe how drugs are absorbed by the body.
- Describe how drugs are distributed by the body.
- Describe how drugs are metabolized by the body.
- Describe how drugs are excreted by the body.
5.1. Overview of Pharmacology
Section Learning Objectives
- Differentiate between pharmaceutics, pharmacokinetics, and pharmacodynamics.
- Define the four different components of pharmacokinetics.
As mentioned in the introduction, this chapter is the start of our exploration of pharmacology, which is the study of the actions and effects of drugs. You can easily see how such a field is relevant to a class about drugs and behavior. Pharmacology is one of the basic biomedical sciences that form the foundation of many health sciences and is critical to understanding therapeutic drugs and drug treatments.
5.1.1. The Pharmaceutical Sciences
The Pharmaceutical Sciences constitute an interdisciplinary field that centers on the discovery, development, formulation, and application of new chemical entities to the treatment of physical and mental disorders.
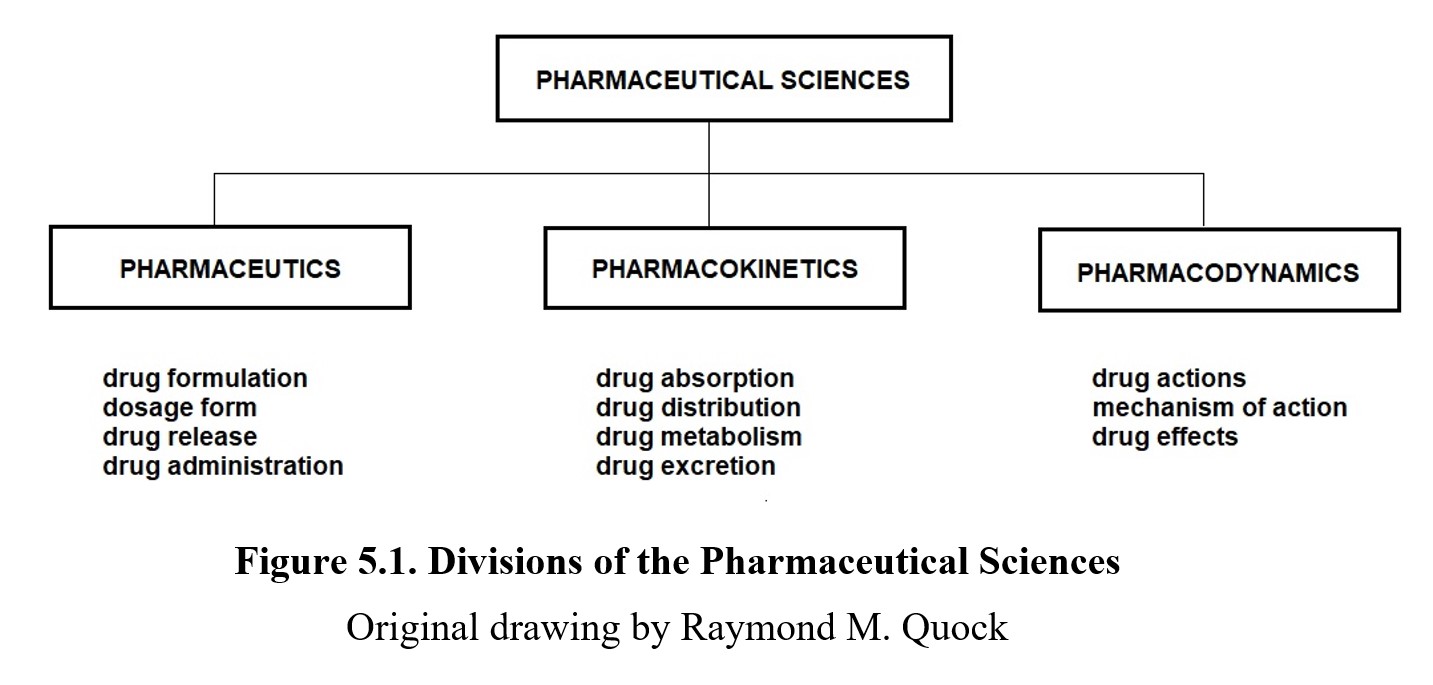
Pharmaceutics is the study of how a drug is formulated and dispensed. In the past, pharmacists often dispensed drugs directly as a powder containing just the active ingredients. Nowadays, drugs are usually designed with a dosage form in mind, which is a mix of active and inactive ingredients prepared in a particular form, such as a tablet, capsule, or liquid. The particular dosage form and optimal route of administration (e.g., oral, injection, nasal spray, transdermal patch) allow for greater control over the dose of the drug that is taken.
Pharmacology can be divided into two main branches: pharmacokinetics, which is the study of how the drug moves around the body, and pharmacodynamics, which is the study of how the drug changes the body. You can use these meanings to tell the two terms apart; the suffixes ‑kinetics [movement] and ‑dynamics [change] refer to how the drug moves and what the drug changes.
Pharmacology is only one of many different areas of study related to drugs. Another example is medicinal chemistry, which is the synthesis of new drug compounds. We briefly touched on it during the discussion of the New Drug Approval process in Chapter 1, although not by name. There are other fields as well, each with many different subspecialties.
Although we will not cover pharmaceutics in detail in this course, it is worth knowing because of the relationship between pharmaceutics and pharmacokinetics. As you can see in the figure above, the dosage form determines how the drug is made available to the body. This has relevance to pharmacokinetics of the drug, which, in turn, influences the pharmacodynamics of the drug.
5.1.2. ADME
The focus of this chapter is pharmacokinetics, which is concerned with how the drug moves throughout the body. In reality, the drug isn’t moving on its own—it’s being moved around by the natural systems in the body. Because of this, we can also say that pharmacokinetics is what the body does to the drug. (Next chapter we will look at pharmacodynamics, which is the converse—what the drug does to the body.)
There are four main things that the body does to the drug: it absorbs it into the bloodstream, distributes it to various areas of the body, metabolizes it into different compounds, and excretes it from the system. A useful mnemonic that can help you remember this process is ADME—
Absorption
Distribution
Metabolism
Excretion
We will spend the rest of this chapter examining each of these in detail.
5.2. Absorption
Section Learning Objectives
- Define bioavailability and diffusion.
- Explain how a drug’s ability to permeate membranes is critical to absorption and describe what factors can influence this.
- Describe different routes of administration and explain how they influence drug absorption and bioavailability.
The first factor that influences how a drug moves throughout the body is absorption. Absorption describes the movement of the drug from its site of administration to the circulatory system. Drugs may be taken orally, by injection, by inhalation, and other routes. Most drugs will ultimately enter the bloodstream, which will carry or distribute the drug to its sites of action. As such, understanding how the drug gets absorbed into the bloodstream is an important component of pharmacokinetics.
5.2.1. Determinants of Absorption
Following administration, not all drugs will be absorbed into the bloodstream, and not always at the same rate. The amount that does get absorbed is termed bioavailability, expressed as a percentage of the amount administered that enters the circulation. Some drugs will also be absorbed more quickly, which can increase the strength of their effects. Some drugs will be intentionally absorbed more slowly, which can prolong their duration of action.
The bioavailability and rate of absorption depend heavily on how well the drug can diffuse from its site of administration to the bloodstream. Diffusion simply refers to a substance spreading out, i.e., moving from an area of high concentration to low concentration. We have already encountered this idea before when discussing chemical ions and action potentials in Chapter 3. A drug that can pass through membranes easily will diffuse faster than one that cannot.
How well the drug can permeate these membranes depends on certain properties of the drug. Drugs that easily diffuse across cell membranes are small, non-ionized, and lipophilic (from lipo in Greek for “fat” and philos for “friendly). Larger molecules, ionized chemicals, and hydrophilic (from hydro for water) substances all have a harder time passing through membranes. This is because the phospholipid bilayers that comprise cell membranes consist of hydrophilic heads and uncharged tails that repel hydrophilic and ionized molecules.
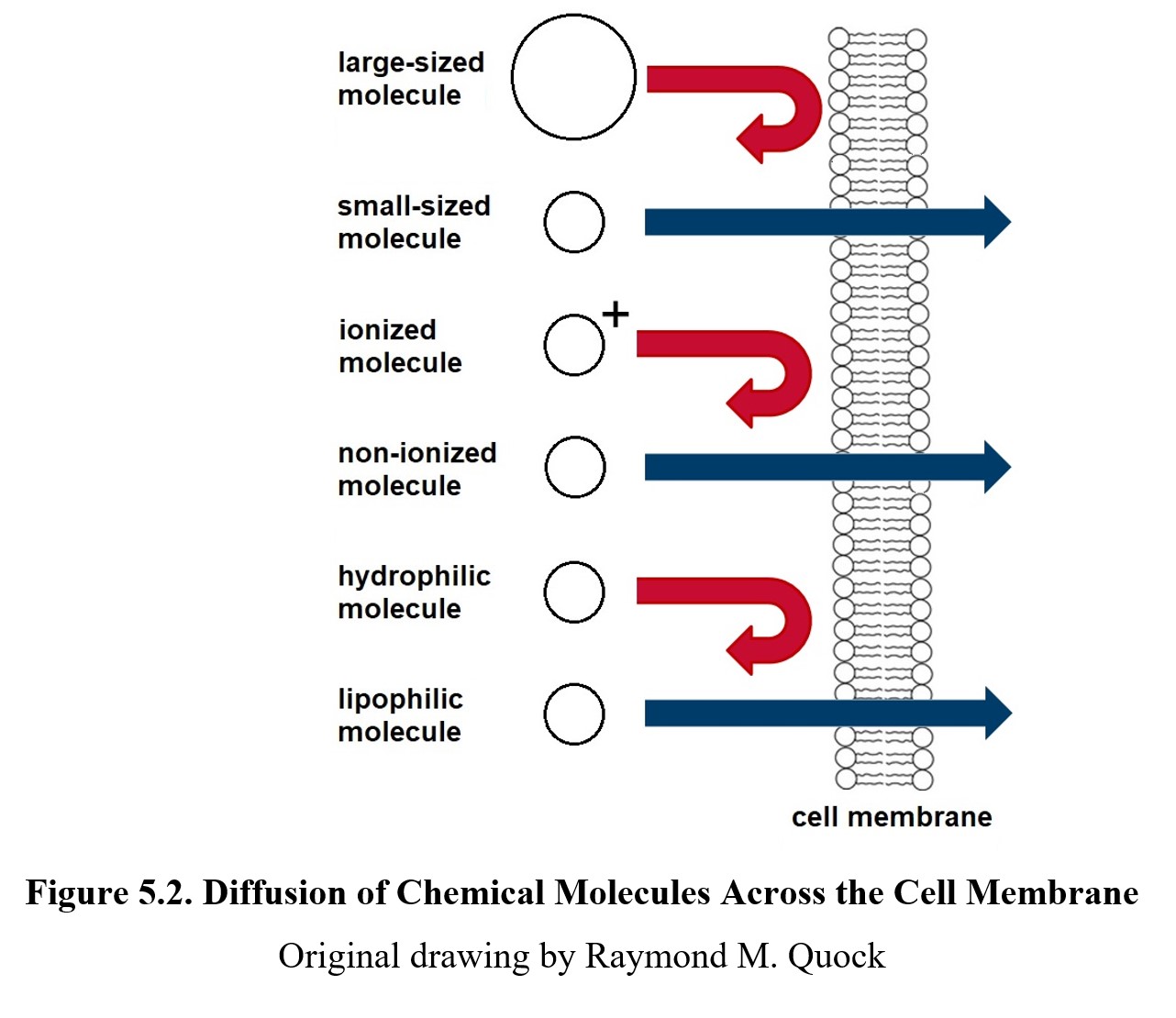
Aside from diffusion that occurs on its own (known as passive diffusion), drugs can also be moved via active transport mechanisms. These mechanisms involving carrier proteins and channel proteins consume energy (ATP) but can move larger molecules and work against concentration gradients. By now, you should be familiar with the ion channels found in nervous tissue; similar channels exist in different cells. Active transport can allow drugs with larger molecules to pass through membranes and be absorbed.
The rate of absorption and bioavailability also depend on the route of administration, or the path that the drug takes to enter the body. Some paths are more direct than others. A drug that is injected directly into the bloodstream will, by definition, have 100% bioavailability because 100% of the administered dose enters the blood for distribution. Drugs that pass through the gastrointestinal tract face a gauntlet of obstacles that will slow down the rate of absorption and reduce the total amount of the drug that reaches the systemic circulation (i.e., the first-pass effect). Recall that the dosage form of a drug can determine how it is taken, so the route of administration is often influenced by the drug’s pharmaceutics. Another way of looking at it is that if a certain route is preferred, the dosage form must be consistent with that route for maximum bioavailability.
For the remainder of this section, we will examine various routes of administration. As we cover each one, pay close attention to how they differ in terms of rate of absorption and bioavailability.
Methods of Drug Administration [7:47]
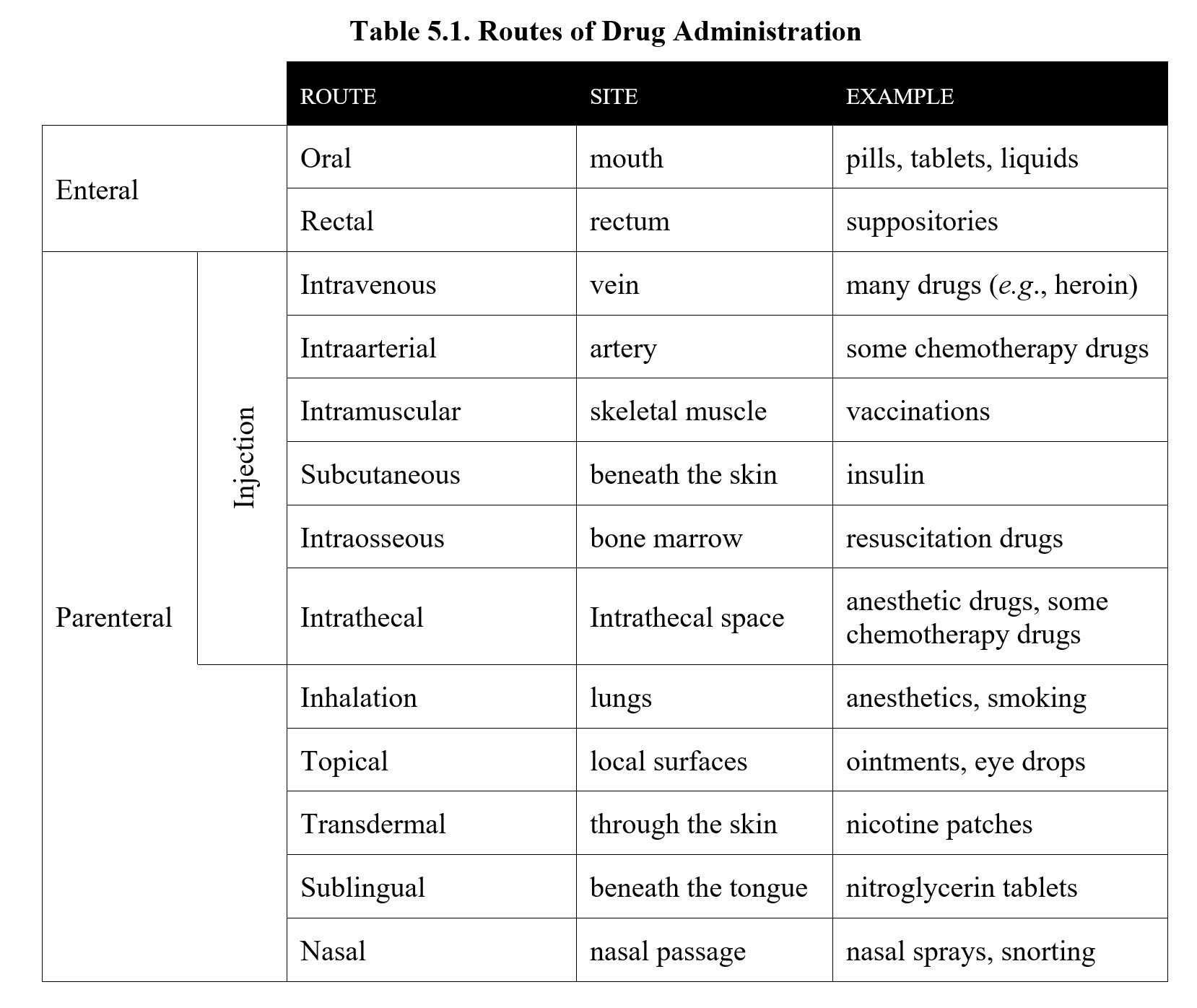
5.2.2. Enteral Routes
There are two general categories of routes of administration. The first type is the enteral route (from enteron in Greek for “intestine”), which refers to the routes that pass through the gastrointestinal or digestive tract. from enteron for “intestine.”). This is usually accomplished through oral administration which has the medical abbreviation p,o, (from per in Latin for “through the” and os for “mouth”). This is the method of drug administration with which you are probably most familiar. Capsules, tablets, and liquids like cough syrups and alcohol are all taken orally although we say we drink alcohol rather than “orally administer” it. Aside from the oral route, there is also rectal administration, which involves inserting the drug directly into the opposite end of the digestive tract as in the case of a rectal suppository or an enema. Of these two routes, the rectal route is faster and simpler.
Drugs taken orally must first pass through the stomach on the way to the intestines. The stomach typically absorbs drugs more slowly than the intestines, so it can take longer for the drug to be absorbed. If the stomach is full of food, the drug will spend more time in the stomach, reducing the rate of absorption even further. In the chapter on alcohol, we will understand the adage “don’t drink on an empty stomach.” Finally, the dosage form of oral medication is important, because not all drugs can survive the highly acidic environment of the stomach. These drugs must be enclosed in acid-resistant capsules that delay the release of the drug until after it reaches the intestines.
Both oral and rectal routes pass through the intestinal walls which are comprised of epithelial cells. Drugs must be able to permeate these cells in order to be absorbed. Otherwise, they will simply pass through the intestines and be excreted in the feces without accomplishing anything. If a drug cannot be absorbed through the intestinal wall, it will require a different route of administration altogether.
Even if an orally administered drug makes it through the intestinal walls and into the bloodstream, it will be taken to the liver before circulating to the rest of the body. This is significant because the liver often metabolizes drugs which may reduce the bioavailability further. We will cover this in detail when we reach the section on metabolism. For now, it is enough to grasp that the oral route tends to have lower bioavailability. In spite of this, taking medication by mouth is generally the most convenient option.
5.2.2. Parenteral Injection Routes
The alternative to the enteral is parenteral, (from para in Greek for “beside” and énteron for “intestine”) which includes all the routes that do not pass through the gastrointestinal tract. This often involves an injection of some sort, although there are non-injection parenteral routes as well. We will start by first looking at the routes that involve injection of the drug.
First is intravenous (i.v.), which involves injecting the drug into a vein. Because the drug is injected directly into the bloodstream, i.v. administration is fast and results in 100% bioavailability. For drugs like heroin, this manifests as an immediate rush of pleasure, which is why they are often administered this way. Intravenous therapy is also ideal for emergency use in hospitals, as it can be used for blood transfusions, fluid replacement, parenteral nutrition, and medications. The downside of the i.v. route is that it requires skill and knowledge to use since a vein must be found and pierced with a needle. Although some abusers of drugs like heroin become proficient at i.v. injections, veins can collapse if they are penetrated excessively. Another complication of the i.v. route is that, once the injection is completed, the drug cannot be recalled in the event of inadvertent administration of an excessive dose.
Intraarterial (i.a.) is another injection route into the circulatory system. This alternative route is used in the case of delivering higher doses of chemotherapeutic agents into the carotid artery for treatment of brain tumors.
Another common method of injection is intramuscular (i.m.) The name suggests medications are injected into the skeletal muscle such as the gluteus maximus which is the safest site for i.m. injections. From the muscle, the drug is absorbed into the bloodstream. The i.m. route results in high bioavailability but is somewhat slower than i.v. Although many different drugs can be administered intramuscularly, most people have likely experienced i.m. administration when receiving a flu shot or COVID-19 vaccination.
Aside from injecting the drug into the veins or muscles, it can also be injected below the skin, known as subcutaneous (s.c.). Compared to the i.m. or i.v. routes, s.c. absorption takes longer because there are fewer blood vessels underneath the skin. In exchange, s.c. injections are good for drugs that need to be absorbed for a long period, which is why insulin is usually administered by this route. Another reason for s.c. administration of insulin is that orally administered insulin is a protein that is broken down in the digestive tract.
Another method is an intraosseous (i.o.), which involves injecting directly into the bone marrow. As you may recall from biology, the marrow is the part of the bone that is responsible for producing new blood cells, and, as such, has direct access to the bloodstream. Intraosseous infusion is comparable to i.v. in that it delivers drugs and fluids directly into the circulatory system. It is also comparable to i.v. in terms of speed of absorption and bioavailability. Intraosseous is useful when i.v. access cannot be established quickly, such as with trauma patients or during cardiac arrest. An example of a drug that can be administered in such cases is epinephrine used to resuscitate patients.
The last injection route we will look at is intrathecal (i.t.), which means injecting into the theca or sheath of the spinal cord that contains the cerebrospinal fluid. This route is notable because it bypasses the blood-brain barrier, which is an impediment to drug distribution that we will cover in more detail later in this chapter. Certain anesthetics and anticancer drugs are administered in this manner for spinal anesthesia and the treatment of brain tumors.
5.2.3. Parenteral Non-Injection Routes
Now we will look at routes that bypass the gastrointestinal tract without the need for a needle. First up is inhalation, which involves inhaling the drug as a vapor. This produces high bioavailability like i.v. administration but is actually faster because the drug enters the circulatory system in the lungs, instead of at the veins where it has to be carried back to the heart before being distributed via the circulatory system. This makes inhalation a common method for recreational drug use, as it provides an immediate effect. Although smoking is convenient, the chemical byproducts produced by it can damage the lungs. Safer methods of inhalation are found in therapeutic drugs, such as asthma inhalers that contain corticosteroids or bronchodilators or the general anesthetics used during general surgery.
Another method is topical, which means applied to a certain place, often a body surface. This is typically the skin, as in the case of ointments or creams, but can also refer to eye drops and eardrops. Topical administration does not result in systemic effects. That is, instead of being absorbed in the bloodstream and distributed to the rest of the body, the doses of topical medications simply work locally at their intended site of action close to the site of administration.
By comparison, transdermal administration (meaning “through the skin”) does result in the drug reaching the systemic circulation, as higher concentrations of drug are gradually absorbed by capillaries in the skin to enter the bloodstream. This process is very slow and is determined by a membrane that controls the rate of absorption. Transdermal is similar to s.c. in that it can support sustained absorption of the drug. Examples of drugs that are administered transdermally include nicotine for smoking cessation, fentanyl for management of chronic pain, scopolamine for treatment of motion sickness, and others. See the description of a drug patch below:
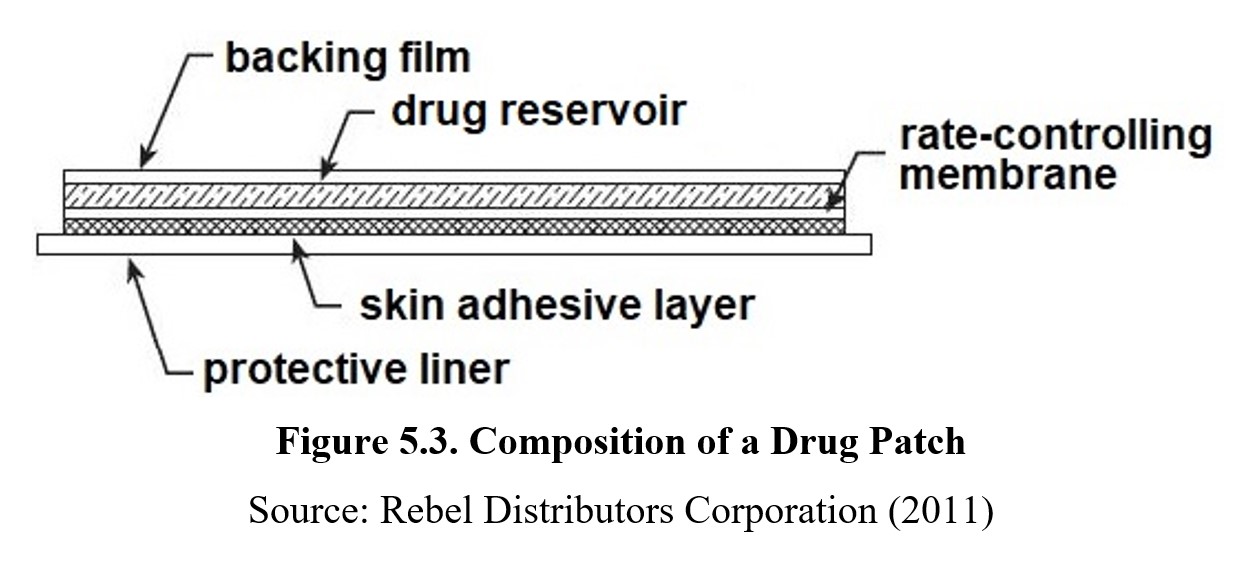
A similar method of administration is sublingual (from sub in Latin for “under” and lingua for “tongue”.). This method involves placing a tablet underneath the tongue, where it dissolves and is absorbed by the capillaries there. Sublingual medications can also be applied as dissolvable strips or liquid drops. Many people are familiar with nitroglycerin tablets, which are typically administered sublingually to treat angina pectoris.
Finally, drugs can be administered through a nasal route. The nasal passage contains mucosal membranes that can absorb drugs into the capillaries, similar to sublingual or transdermal routes. Drugs can be applied as a liquid or powder, the latter of which dissolves inside the nasal passage. Examples of drugs that use this route are nasal decongestant sprays and some recreational drugs that are snorted (most notably cocaine).
Before moving on, take a moment to look over the table summarizing each of the routes of administration below.
Here is a short video that uses aspirin as a representative drug followed by another video reviewing the main points of pharmacokinetics.
Aspirin Journey Through the Body [3:49]
Pharmacokinetics: How Drugs Move Through the Body [7:54]
5.3. Distribution
Section Learning Objectives
- Explain plasma protein binding and how it affects drug distribution.
- Describe the blood-brain barrier and explain how it affects drug distribution.
Once the drug enters the circulatory system, the bloodstream carries it to the site of action. This process is known as distribution. Distribution determines how much of the drug reaches the site of action, similar to how absorption determines how much enters the bloodstream in the first place. In a manner of speaking, the circulatory system is akin to the freeway system that will let you reach your destination. Absorption is the on-ramp to the freeway. If the on-ramp is metered, it only allows cars to enter the freeway at a limited rate. In this section, we will examine two factors that influence drug distribution: plasma protein binding; and the blood-brain barrier.
5.3.1. Plasma Protein Binding
Not all of the drug that is absorbed will be free to activate receptors in the target area. Some amount of drug may be retained in the blood, unable to diffuse out of the circulatory system to the site of action. This is because the plasma in our blood contains many different proteins, some of which can reversibly bind to drugs in a process known as plasma protein binding. View the following video from Dr. Steven Farmer on significance of plasma protein binding.
Let us review the information covered in the video. The drug-binding plasma proteins act like sponges, “soaking” up the drug by binding to it. Once bound to the protein, the drug will be stuck inside the circulatory system and unable to reach the site of action. To activate receptors, it is necessary to first saturate the protein binding sites in the blood, meaning a larger amount of drug is required. The amount depends on how well the drug binds to the proteins. A drug that has a binding rate of 99% means that only 1% of the administered drug is responsible for the subsequent therapeutic or adverse drug effects.
What complicates this process is that other drugs may also compete for these binding sites as well. If a new drug that binds to the same sites is introduced, it will displace some of the original drug, increasing the amount that reaches the target area. The magnitude of the change depends on each drug’s ability to bind to the plasma proteins. If a drug has a high binding rate (like in the 99% example above), even a small change in the amount of available binding sites can double or triple the amount of drug that reaches the site of action, which can result in adverse effects. One of the best examples is that of aspirin displacing the oral anticoagulant warfarin from plasma proteins and causing excessive bruising or bleeding. The opposite is also true: discontinuing a drug can render another drug ineffective, as seen in the example provided in the video. This is why it is dangerous to drink alcohol with some medications and why physicians need to be aware of what medications you’re taking before prescribing a new one.
5.3.2. The Blood-Brain Barrier
Due to how important our brain is, our body has an extra layer of security meant to protect it from pathogens and toxins that may be carried in the blood. This defense is called the blood-brain barrier (sometimes abbreviated BBB), and it is an additional boundary that separates the circulatory system from the brain. Watch this video from 2-Minute Neuroscience that explains what it is and how it works:
2-Minute Neuroscience: Blood-Brain Barrier [1:59]
As mentioned in the video, the blood-brain barrier is formed by tight junctions of endothelial cells, which are the cells that line blood vessels. Unlike in most parts of the body where there are gaps between the cells to let substances through, the tight junctions in the blood-brain barrier limit which substances can diffuse through the capillaries. These tight junctions are formed with the help of astrocytes which are a type of glial cell found in the brain. Recall from Chapter 3 how glial cells are the support cells of the nervous system).
The blood-brain barrier helps maintain a constant environment for the brain and protects it from foreign substances or neurotransmitters from other parts of the body. Consequently, not all drugs can pass through the barrier. Recall the discussion of a drug’s ability to permeate membranes in the previous section. Large, ionized, or hydrophilic drugs will find it harder to reach the brain. This means that if a drug’s intended site of action is in the brain, it needs to find some way of penetrating the blood-brain barrier, such as by being lipid-soluble or relying on active transport mechanisms.
Similar barriers exist in other parts of the body. The cerebrospinal fluid is protected by a barrier that lets in some substances that are blocked by the blood-brain barrier. The placental barrier also exists between a fetus and its mother, although this barrier is much more permeable to drugs and other substances, which is why expectant mothers are advised to abstain from drinking, smoking, or other drug use as the drugs can cross the barrier and harm the fetus.
5.4. Metabolism
Section Learning Objectives
- Explain the first-pass effect and how it affects the bioavailability of oral drugs.
- Describe the metabolic processes that occur in the liver and explain the role of enzymes such as cytochrome P450.
- Explain how enzyme inducers and inhibitors affect bioavailability.
- Describe prodrugs and explain why they are useful.
The human body is not limited to simply moving drugs around. After all, many substances that we consume, intentionally or not, can be toxic. Our bodies can chemically modify these substances in a process known as metabolism or biotransformation. Metabolism can transform inert substances into nutrients or alter toxic chemicals so that they are more easily expelled from the body.
When a drug is metabolized by our body, the resulting compound is called a metabolite or by-product. If a metabolite has a physiological effect of its own, it is called an active metabolite. However, sometimes drugs are transformed into inactive metabolites that do not affect the body. The transformation of a drug into active or inactive metabolites affects all other aspects of pharmacokinetics, which is why we will be taking a closer look at the process in this section.
5.4.1. Metabolism in the Liver
The main organ of metabolism in the body is the liver. Although biotransformation can occur in other tissues, we will focus on the liver for this course. The reason why the liver is so significant is that everything we eat and drink is sent to the liver first for processing. Substances are absorbed from the intestinal tract and carried directly to the liver by the hepatic portal vein. It is important to note that 75% of the hepatic blood supply comes from the hepatic portal vein. Thus, orally ingested drugs are only able to reach other parts of the body after passing through the liver (see image below).
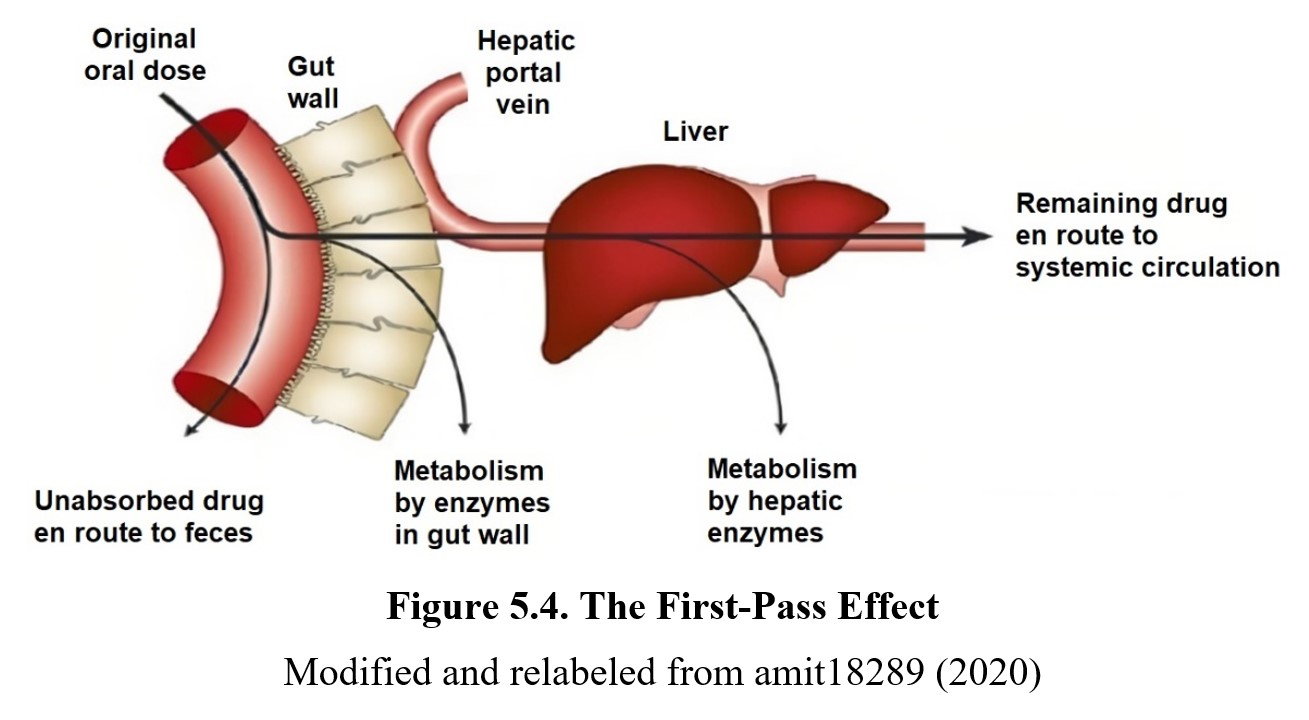
What this means is that drugs absorbed from the intestinal tract are taken straight to the liver before they can be distributed to the site of action. This is known as the first-pass effect or first-pass metabolism, where some drug is immediately metabolized in the liver before reaching the systemic circulation. This reduces the bioavailability of drugs that are orally administered but not drugs administered by other routes. First-pass metabolism also affects rectal administration but to a lesser degree, as some of the drugs can enter systemic circulation right away.
Bioavailability and First Pass Metabolism [4:38]
(N.B. Don’t worry about all the math that starts around 3:00 unless you’re a math major.)
What exactly does metabolism entail? All metabolic processes are chemical reactions aided by enzymes, which are proteins that catalyze (speed up) the reaction. Metabolic reactions are classified into two groups: phase I and phase II. Phase I reactions typically transform the drug to make it more hydrophilic through oxidation, reduction, or hydrolysis. This is necessary because it is difficult to eliminate lipid-soluble molecules from the body, so the liver alters them to be hydrophilic or water-soluble instead. Most reactions in this phase involve enzymes that belong to the Cytochrome P450.family of enzymes.
What is cytochrome P450?
Cytochrome P450 enzymes belong to a superfamily of enzymes that are involved in phase I reactions. Cytochrome P450 enzymes are often abbreviated as CYP. “P450” comes from the peak absorption band at 450 nm of the enzyme when complexed with carbon monoxide. The nomenclature for CYP P450 enzymes is complicated. Individual enzyme names typically include a number for the gene family, a capital letter for the subfamily, and another number for the individual gene. Examples include CYP3A4 and CYP2D6. As illustrated below, more than 75% of phase I reactions are catalyzed by enzymes that begin with the CYP prefix. You will encounter more CYP enzymes as we examine certain drugs that are metabolized by CYP enzymes.
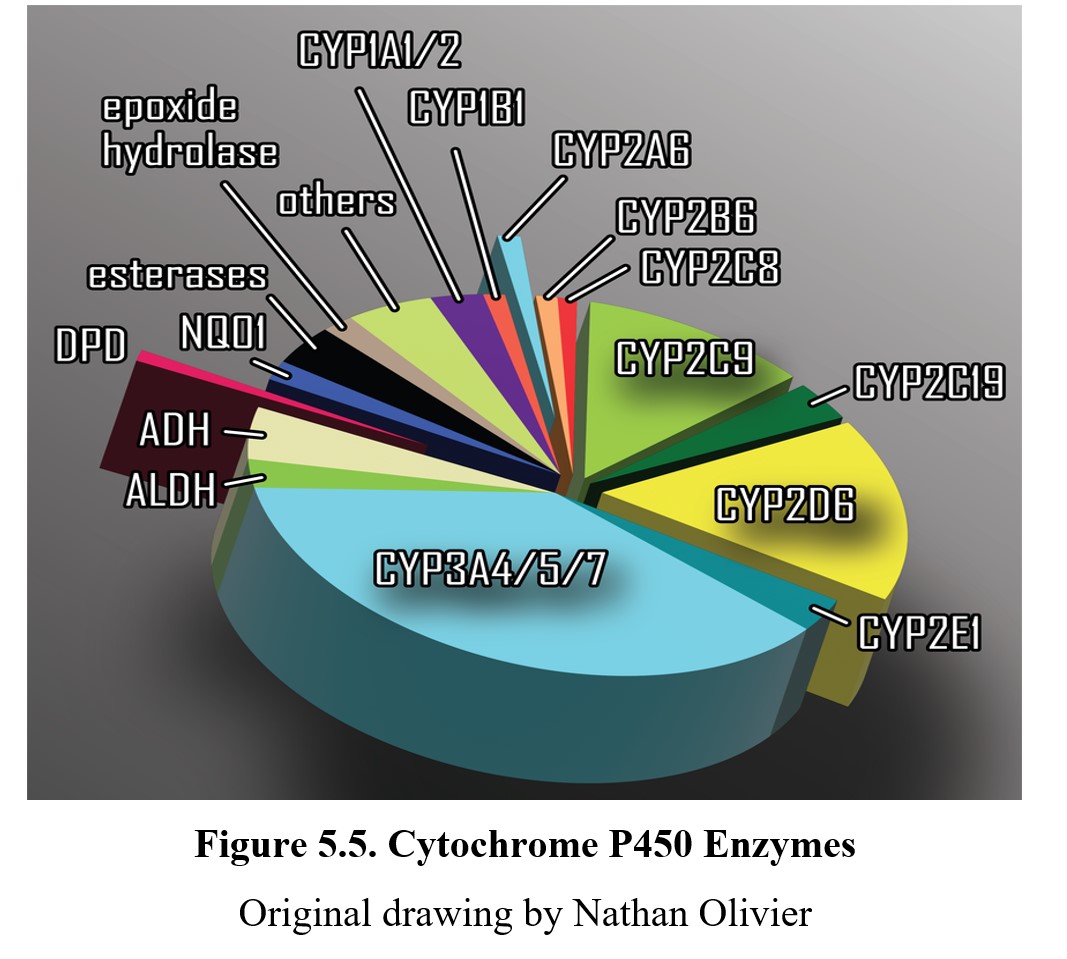
Figure 5.5. Cytochrome P450 Enzymes
Original drawing by Nathan Olivier
It would be incorrect to assume that the role of CYP P450 enzymes is solely involved in breaking down or inactivating drugs. CYP P450 enzymes are essential in the synthesis of steroid hormones, cholesterol, and eicosanoids.
This video reviews drug metabolism, enzyme induction, enzyme inhibition, and prodrugs. (Don’t worry about the names of the specific CYP P450 enzymes.)
Some drugs and metabolites also undergo phase II reactions, which attach polar groups such as sulfate or glucuronic acid to the drug molecule in a process known as conjugation. These polar groups make the molecules even more hydrophilic, ensuring that they can be more easily excreted. The enzymes that make this possible are transferase enzymes such as methyltransferase, glucuronosyltransferase, and acetyltransferase. Cytochrome P450 enzymes are not involved in phase 2 reactions.
5.4.2. Enzyme Induction and Inhibition
Because enzymes control the rate at which drugs are metabolized, changes in enzyme activity have a significant impact on drug bioavailability. If there is increased expression or production of an enzyme (induction), the metabolism of the drug will speed up, and less of the drug will be available to produce its effects. Drugs that increase the expression of enzymes are enzyme inducers.
Enzyme inducers can come from substances other than drugs. Saint-John’s wort, which is often used as an herbal remedy, induces the enzyme CYP3A4. This can reduce the effectiveness of drugs that are metabolized by CYP3A4, such as indinavir, an anti-HIV drug. Some drugs even induce the very enzymes that metabolize themselves. Phenobarbital, a barbiturate used to treat epilepsy, is one such example. Over time, repeated administration will result in the drug having a reduced effect due to it being metabolized at a faster rate. This is one mechanism for the development of tolerance to barbiturates
Drugs can also act as enzyme inhibitors by reducing the expression of the enzyme or blocking the enzyme’s active sites. As you might expect, this slows the metabolism of the drug, increasing its bioavailability and prolonging its effects. As with inducers, this can be done by the drug itself or by another substance. A notable example is grapefruit juice. Grapefruit juice contains compounds that inhibit CYP3A4 enzymes which can increase the concentrations of many medications that are metabolized by CYP3A4. Selective serotonin reuptake inhibitors (SSRIs) are also notable for inhibiting various CYP enzymes.
If these concepts are still confusing to you, you may want to review them before you move on by watching this short video:
Enzyme Inhibition and Enzyme Induction [3:19]
5.4.3. Prodrugs
There is one final concept worth discussing in the context of drug metabolism. So far, we have framed metabolism as a process that tends to negatively impact the drug. Although that is the case for most drugs, not every biotransformation reduces the effectiveness of the drug. The metabolite can be more pharmacologically active than the drug initially administered. This is the case for prodrugs. These are drugs that are administered in an inactive or less active form that only becomes active or more active after the drug is metabolized.
Why are prodrugs useful? The chemotherapy drug 5-fluorouracil (5-FU) mentioned in the video is a good example. Sometimes the active form is too toxic to be administered directly. There are other potential reasons as well. Some prodrugs are better at crossing cell membranes. If you recall from the previous chapter, Parkinson’s disease is caused by a deficiency of dopamine. It would be nice to administer dopamine directly to treat the disease, but dopamine cannot cross the blood-brain barrier. Fortunately, its precursor, L-DOPA, can pass the barrier instead. This allows it to be administered as a prodrug that is converted to dopamine in the brain.
5.5. Excretion
Section Learning Objectives
- Describe various routes of excretion for drugs.
- Differentiate between first-order and zero-order elimination kinetics
- Define the half-life of a drug.
The final step in every drug’s journey is to leave the body in some manner. Excretion is the elimination of a drug from the body, either in its unchanged form or as a metabolite. Although this may seem like an automatic process, it cannot be taken for granted. If drugs or other waste products accumulate in the body, they can cause harm, which is why energy must be constantly spent removing these substances from the body.
5.5.1. Routes of Excretion
Although drugs can be excreted through a variety of routes, most drugs are excreted by the kidneys into the urine. The kidneys work like filters, removing the waste products from the bloodstream. The exact process is complex and beyond the scope of this class, but it involves the same process of diffusion mentioned in the absorption section. Recall that lipophilic drugs need to be metabolized into hydrophilic metabolites before they can be filtered out by the kidneys since lipid-soluble molecules would simply reenter the bloodstream. Nonionized molecules are also difficult to excrete for the same reasons. The kidneys are also unable to excrete any drug that is still bound to plasma proteins.
Because most drugs are excreted through the kidneys, drug tests usually involve taking a urine sample. By measuring the presence of a drug or its metabolites in the urine, it is possible to determine whether a drug was recently present in a person’s body. Drugs can also be excreted through the liver as bile or feces, through sweating, and even through tears, although these routes are less important when it comes to drugs. Drug tests that involve mouth swabs are testing your saliva, which is another excretory route. Another type of drug test that you have probably heard of is the breathalyzer, which measures alcohol levels in the blood just by breathing into it. This is possible because the lungs can expel certain drugs like alcohol or anesthetics from the bloodstream directly.
There is one final route that requires consideration: mother’s milk. Some drugs can be eliminated through mother’s milk, which could have adverse effects on breastfed infants. Although cases are rare, special caution is warranted as infants have livers and kidneys that are still developing, making them more susceptible to any toxic effects.
5.5.2. Elimination Kinetics
As a drug is eliminated from the body, the amount of drug remaining decreases over time. If we were to graph how much drug remains in the bloodstream, for most drugs we would get a hyperbolic shape like that shown in the graph below:
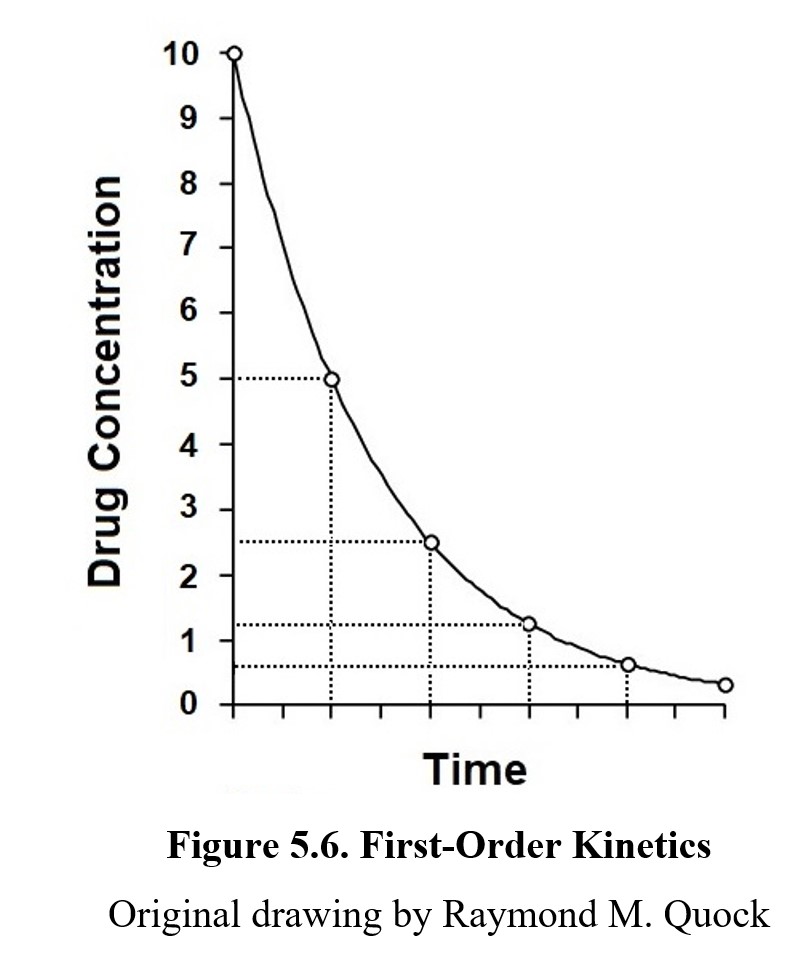
This curved shape follows first-order kinetics, which means it is eliminated at a rate proportional to the amount of the drug. More specifically, first-order kinetics refers to a drug being eliminated in half-lives. A half-life is the amount of time that must pass for the body to eliminate half of the drug. In the figure above, the vertical dotted lines delineate half-lives of the drug. Take methadone, a drug used in therapy for patients with opioid addiction. In a typical patient, the half-life of methadone is approximately 24 hours (Berkowitz, 1976). If we start with 80 mg of methadone in the bloodstream, after 1 day has passed there will be 40 mg remaining. After 2 days, 20 mg; after 3 days, only 10 mg, and so on. Under first-order kinetics, the drug will be eliminated rapidly at the start, but the rate will taper off as the concentration of the drug in the blood decreases. The elimination of most drugs will follow first-order kinetics.
Not all drugs follow this pattern, however. Some drugs, such as alcohol, are eliminated at a constant rate. The amount of the drug eliminated is always the same no matter how much of the drug there is. This is known as zero-order kinetics. If we were to plot the concentration of a drug being eliminated under zero-order kinetics, it would look like the graph below:
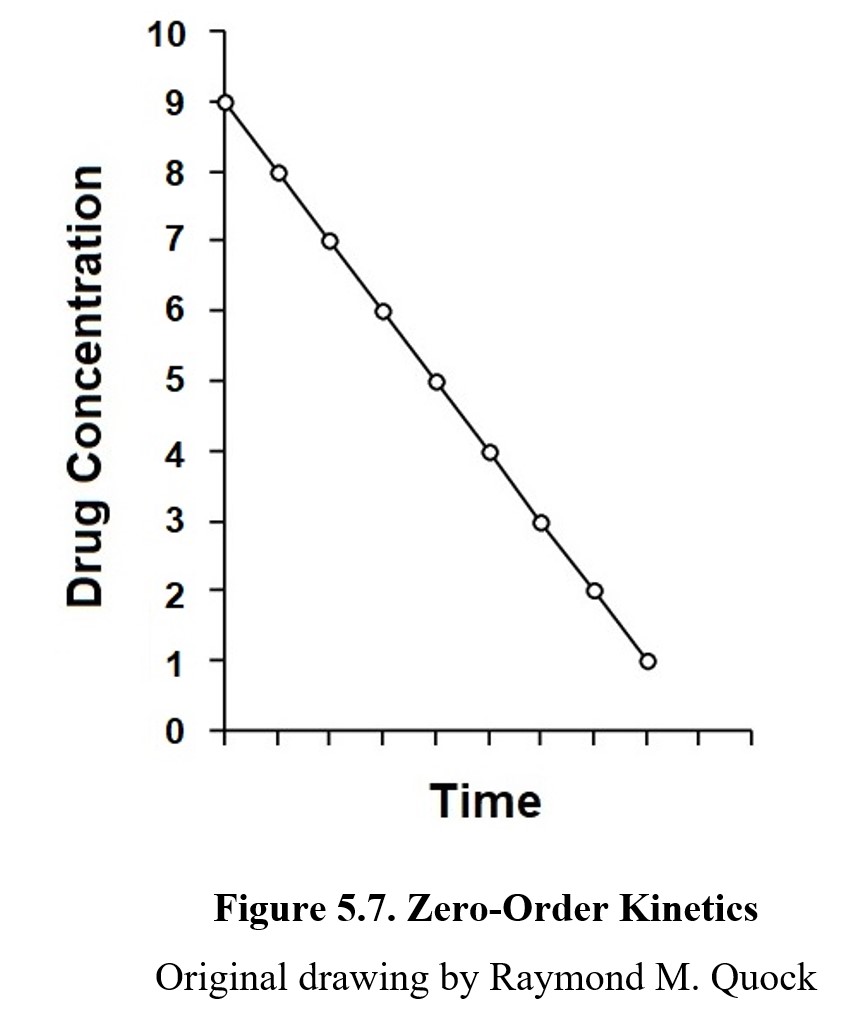
Because the drug is eliminated at a constant rate, the slope of the graph is a straight line instead of a hyperbolic curve. A few drugs such as aspirin follow first-order kinetics for the most part, but once a high enough concentration of the drug is reached, the enzymes that metabolize them become saturated. When the enzymes become saturated, increased metabolism of the drug is impossible, and the elimination rate follows zero-order kinetics instead. This is rare to see in practice, however, because such levels of concentration are well above therapeutic levels.
Chapter Summary and Review
In this chapter, we explored pharmacokinetics. After providing an overview of the different branches of pharmaceutical sciences, we took a journey through the four components of pharmacokinetics captured in the acronym ADME—absorption, distribution, metabolism, and excretion. We discussed factors that affect absorption and compared different routes of administration. We learned about plasma protein binding and the blood-brain barrier and how they influenced drug distribution. We examined metabolic processes, cytochrome P450 enzymes, and prodrugs. Finally, we covered the pathways and rates at which drugs can be excreted from the body. This was a dense chapter, so make sure to give yourself time to digest it, and do not forget to check your understanding with the practice questions below.
Chapter 5 Practice Questions
Answer the following questions:
- What disciplines make up the pharmaceutical sciences?
- What do pharmaceutics, pharmacokinetics, and pharmacodynamics study?
- Distinguish between enteral and parenteral routes of drug administration?
- Rank these routes of administration from fastest absorption to slowest: intramuscular injection, inhalation, and transdermal.
- What is the bioavailability of a drug that is administered intravenously?
- Explain the first-pass effect. Which routes of administration are subject to the first-pass effect?
- Which drug is more sensitive to competition for plasma protein binding sites: one with a high rate of binding, or one with a low rate of binding?
- Describe what makes up the blood-brain barrier.
- How does a cerebral blood vessel differ from a typical blood vessel?
- What is cytochrome P450?
- Does enzyme induction increase or decrease the bioavailability of a related drug? What about enzyme inhibition?
- What is a prodrug, and in what circumstances would it be useful?
- Would a lipid-soluble molecule be able to be excreted by the kidneys? Explain why or why not.
- When does a route of excretion become a route of administration?
2nd edition
